


染色体平衡易位是染色体结构变异常见的一种类型,正常人群平衡易位携带者发病率约为0.2%。染色体平衡易位携带者由于没有存在遗传物质减少或增加,临床可表现为正常,但在生育下一代时由于染色体重新分配,可产生遗传物质正常、缺失、重复或与携带者一致的4种配子,其中缺失或重复配子在子代可导致流产、死胎、严重出生缺陷的胎儿出生[1]。本文从一例父源性遗传导致非平衡易位患儿的基因型与表型进行分析,从两者相关性进行遗传学研究,为遗传咨询提供相关依据,现报道如下。
对象与方法
一、研究对象
2018年6月14日我科接诊一例出生7 d因宫内窘迫剖宫产娩出早产儿,双手多指畸形(六指),为确定患儿是否存在遗传性疾病,进行家系染色体核型分析及患儿全基因组单核苷酸多态性微阵列分析技术(SNP-array)检测,并收集分析其病史,实验室及辅助科室相关资料,所有受检者对上述检测知情并签署知情同意书。本研究已获得本院伦理委员会审核批准(批件号:CR2023-008-01)。
二、染色体核型分析
采集患儿及父母肝素钠抗凝外周血进行细胞培养,72 h后加入秋水仙素终止细胞分裂,通过细胞收获、染色体制备、烤片、显带、吉姆萨染色、显微镜下核型分析确定染色体核型。染色体核型分析计数20个分裂像,分析5个细胞核型,结果参考国际人类细胞遗传学术语命名系统(ISCN 2020)进行核型描述。通过染色体细胞计数及核型分析对染色体数目及结构异常进行诊断,其中染色体数目异常包括多倍体、三体综合征、嵌合体等;染色体结构异常包括易位、倒位、重复、缺失、插入、环状等。
三、基因芯片检测
采用Affymetrix平台750K基因芯片进行全基因组检测分析。检测结果通过查询正常人群的结构变异(DGV)数据库、Decipher数据库、ClinGen数据库、在线人类孟德尔遗传(OMIM)数据库进行注释分析。结果参考美国医学遗传学和基因组学学会(ACMG)指南对拷贝数变异(CNV)的临床意义分级标准分为:致病性(P)、可能致病性(LP)、临床意义不明(VUS)、可能良性(LB)、良性(B)。
四、文献检索
以“t(13;22)”为检索词,对生物医学文献外文数据库(PubMed)、中国知网全文数据库(CNKI)、万方数据库、重庆维普学术期刊数据库进行检索,收集亲代遗传t(13;22)非平衡易位病例进行分析,检索时间截至2023年6月。
结果
一、一例22q13缺失综合征患儿及父母资料
1. 患儿临床资料
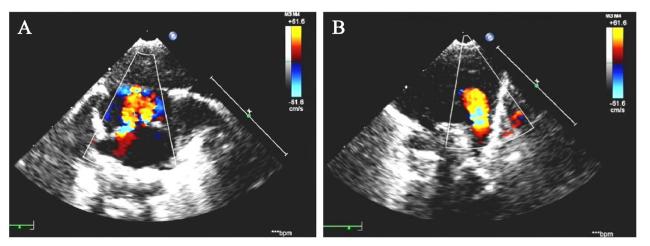
患儿男,出生7 d,系孕36+3周自然受孕早产儿。母亲G1P1,产前未在本院规律产检,孕早期唐氏综合征筛查未见异常,停经14+周外院彩色多普勒超声(彩超)提示胎儿大小约12+4周,颈项透明层厚度(NT)值为2.5 mm。孕中期外院三维彩超未见明显异常。母亲36+3周外院产检发现胎心持续反应差,无下腹部疼痛,阴道无流血、流液,自觉胎动如常,予胎心监护、静脉输液治疗未见改善,遂至我院急诊就诊,拟诊胎儿宫内窘迫,行剖宫产,娩出一男性新生儿。患儿出生时体重2 850 g,身长48 cm,出生后查体反应可,前囟平软,哭声不扬,口唇微绀,双手多指畸形(六指)。辅助检查:血尿粪常规,甲状腺功能,电解质,肝功能等均正常。性激素6项:睾酮3.03 nmol/L,卵泡刺激素< 0.30 IU/L,催乳素1 431.6 mIU/L,孕酮6.89 nmol/L,黄体生成素< 0.07 IU/L,雌二醇88.02 pmol/L。彩超提示:右侧阴囊内睾丸缺如,右侧腹股沟隐睾,左侧睾丸及附睾未见明显异常;肺动脉高压(重度),动脉导管未闭,房间隔缺损(多孔,继发孔型),室间隔小缺损(肌部),三尖瓣反流(轻度),右心房右心室增大,左心室收缩功能正常,见图1。患儿父母非近亲结婚,否认家族遗传病史,无既往病史。
2. 家系染色体核型分析结果
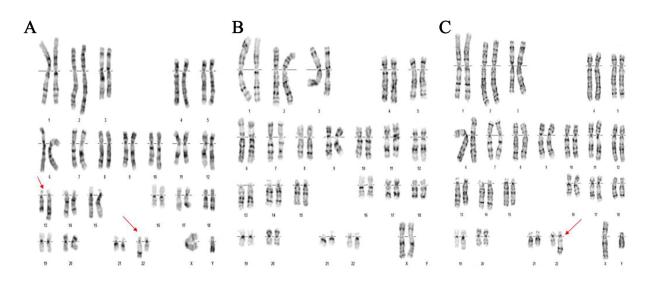
患儿染色体核型分析结果为46,XY,der(22)t(13;22)(q31;q13.3),22号衍生染色体由13号染色体与22号染色体相互易位产生,13号染色体13q31的远端片段易位到22q13.3处;患儿父亲染色体核型分析结果为46,XY,t(13;22)(q31;q13.3),13号染色体长臂3区1带与22号染色体长臂1区3亚带发生易位;母亲染色体核型分析结果为46,XX,正常核型,见图2。依据以上家系核型结果分析确定患儿22号衍生染色体遗传来源于父源,确定其外周血染色体核型结果为46,XY,der(22)t(13;22)(q31;q13.3)pat。
3. SNP-array检测结果
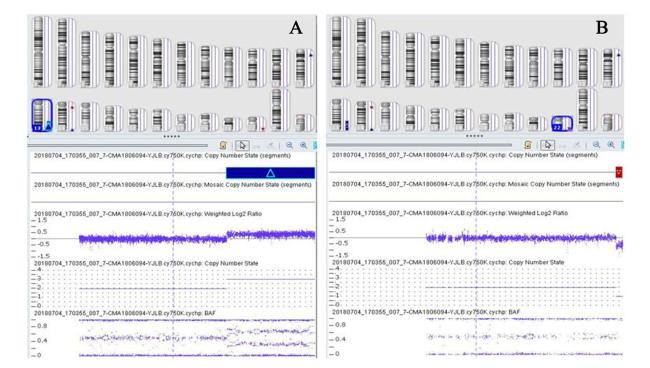
患儿SNP-array结果为arr[hg19]13q31.1q34(79279417-115107733)×3(CNV)、arr[hg19]22q13.33(50059837-51197766)×1(CNV),提示13号染色体长臂13q31.1q34发生35.8 Mb片段重复,22号染色体长臂q13.33发生缺失1.1 Mb片段缺失。见图3。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
arr[hg19]13q31.1q34(79279417-115107733)×3(CNV),检索DGV数据库,片段相似、片段及拷贝数都相似的例数分别为0、0例,该数据库检索结果为不存在片段多态性相似个体。检索Decipher数据库,片段相似、片段及拷贝数相似、片段拷贝数临床表现都相似的例数分别为:3、3、1例,相似病例编号为283913,表型为先天性膈疝,该拷贝数变异片段为新发突变。该数据库检索结果提示存在表型变异相似个体。检索ClinGen数据库,不存在片段变异相似个体。检索OMIM数据库,该片段内包含78个蛋白编码致病基因,其中包含SPRY2、SLITRK1、SLITRK6、MIR17HG、GPC6、TGDS、CLDN10、DNAJC3、ZIC2、PCCA、NALCN、FGF14、ERCC5、SLC10A2、DAOA、LIG4、IRS2、COL4A1、COL4A2、CARS2、ING1、F7、F10、PROZ、GRK1、CHAMP1等基因。根据ACMG指南,综合以上数据库收录病例以及相关临床症状将arr[hg19]13q31.1q34(79279417-115107733)×3(CNV)临床致病性评级为:致病性(P)。
arr[hg19]22q13.33(50059837-51197766)×1(CNV)该片段变异包括已知的遗传综合征区域:22q13缺失综合征[又称费伦-麦克德米德综合征(PMS)],患者通常会出现以下临床表现:常见的发育及行为特征为语言缺乏或严重落后(100.00%)、发育迟缓或智力障碍(91.89%)、肌张力减退(73.17%);轻微畸形特征主要表现为大耳(21.88%)、睑裂狭小(21.88%)、斜视(18.75%),缺失的片段大小与临床表现多样性和(或)严重程度呈正相关[2]。
4. 预后及随访
PMS主要临床表现为智力低下、生长发育迟缓,由于本例患儿年龄较小,临床症状表现不明显;2019年6月6日随访:不会独坐,追视追声可,可逗笑,表情落后,精神发育迟缓;2020年8月22日随访:不会独立行走,后续随访一直未能与家属取得联系,患儿预后情况不详。
二、源于亲代遗传t(13;22)非平衡易位病例文献分析
表1 亲代来源t(13;22)非平衡易位患者的相关文献统计 |
| 第一作者 | 遗传来源 | 患者核型 | 患者临床表现 |
|---|---|---|---|
| Moedjono S J[4] | 母方 | 47,XY,+der(13),t(13;22)(q12;q13) | 身材矮小、颈部短小、胸呈盾形和智力低下 |
| Kim H J[5] | 母方 | 47,XY,+der(22),t(13;22)(q22;q12) | 面部畸形,关节屈曲,喂养困难,癫痫,房间隔缺损,出生11 d因败血症死亡 |
| Mutchinick O[6] | 母方 | 47,XY,+der(22),t(13;22)(q22;q11) | 小头畸形,发育迟缓,智力低下,双侧隐睾,癫痫 |
| 本例患儿 | 父方 | 46,XY,der(22),t(13;22)(q31;q13.3) | 多指畸形(六指),右侧腹股沟隐睾,动脉导管未闭,房间隔缺损,精神发育迟缓 |
讨论
本研究患儿的染色体核型为46,XY,der(22)t(13;22)(q31;q13.3)pat,通过家系核型分析为父源的染色体平衡易位再次分配导致,其中13号染色体长臂13q31.1q34发生35.8 Mb片段重复,检索DGV数据库未发现存在片段多态性相似个体;检索Decipher数据库提示存在表型变异相似个体,其中片段相似3例、片段及拷贝数相似3例、片段拷贝数及临床表现都相似1例,相似病例的临床表现为先天性膈疝。ClinGen数据库不存在片段变异相似个体。OMIM数据库中,该片段内包含78个蛋白编码致病基因,其中与本研究患者临床表型相关的基因分别为DZIP1、NALCN。因此,该片段可能为致病性(P)。另外,患儿22号染色体长臂q13.33发生1.1 Mb片段缺失,该片段变异包括PMS区域。
DZIP1基因是位于13q31.3-32.1的常染色体显性基因,Toomer等[9]对二尖瓣脱垂3(MVP3)的4代家系进行了全外显子组测序(WES),并鉴定了DZIP1基因(608671.0001)错义突变的杂合性,发现了DZIP1亚型存在于S70R和S24R中,该突变在家族中与疾病完全分离,主要临床表型为双小叶脱垂并二尖瓣反流,本研究患儿心脏彩超提示轻度三尖瓣反流与MVP3临床表现相似。Bourque等[10]在一例自然受孕早产儿中发现了NALCN基因中一个纯合突变c.3910C>T,临床表现为早产、呼吸窘迫、癫痫、隐睾、生长发育落后、语言能力迟缓,与本研究患儿呼吸窘迫、早产、隐睾等临床表现相似。
目前PMS尚无标准诊断流程,临床诊断需与肌张力低下、发育迟缓、言语迟缓和(或)孤独症等影响相关的综合征(如普拉德-威利综合征、快乐木偶综合征、威廉姆斯综合征、史密斯-马盖尼斯综合征、脆性X综合征、腭心面综合征、孤独症谱系障碍、脑瘫)相鉴别,同时结合分子遗传学技术检测结果进行确诊。本研究通过对该患儿家系细胞遗传染色体核型分析及分子遗传诊断确定患儿PMS遗传模式,为进一步遗传咨询提供依据。PMS不对患者生命造成影响,但由于智力与发育障碍对家庭和社会将产生很大的负担及影响,因此建议染色体核型分析检测应列入婚检常规检测项目,对于有染色体异常的夫妇应选择产前诊断以避免存在出生缺陷的胎儿出生,必要时选择第三代试管技术进行干预,这将有助于降低我国出生缺陷发病率[13]。无创产前检测技术对于13、18、21号染色体非整倍体具有高灵敏度、高特异度的优点,且对染色体微缺失、微重复有一定提示[14]。因此,对于本例13号染色体存在35.8 Mb片段重复的非平衡易位,若早期通过NIPT筛查可能会有异常提示,从而避免出生缺陷发生,这有待后续进一步研究探讨。