


近几年髓鞘少突胶质细胞糖蛋白(MOG)抗体在中枢神经系统炎性脱髓鞘疾病中的作用得到越来越多的关注,已有研究证实其与成人和儿童视神经炎、脊髓炎、脑干脑炎、急性播散性脑脊髓炎(ADEM)样症状密切相关。目前大多数专家认为MOG抗体阳性的中枢神经系统炎性脱髓鞘疾病是一种独立的疾病实体,其临床特点和免疫病理学机制均不同于多发性硬化(MS)和水通道蛋白-4(AQP4)抗体阳性的视神经脊髓炎谱系疾病(NMOSD),因此又被称为MOG抗体相关性脑脊髓炎(MOG-EM)[1,2]。
MOG抗体可表达于成人及儿童患者体内,有研究者认为成人与儿童在临床症状、治疗转归方面有很大不同。笔者对12例MOG-EM儿童患者进行临床研究,发现其一般以意识水平下降、癫痫大发作等ADEM样症状或视力下降起病,预后均良好[3]。目前关于MOG-EM成人患者方面的报道较少,因本病相对少见,在本文中,我们回顾性分析了15例成人MOG-EM患者的临床资料,对其临床症状、影像学特点、实验室检查、治疗及预后情况进行总结分析,以期为深入研究该病提供参考。
对象与方法
一、研究对象
收集2017 年 7 月至 2020年4月于广东三九脑科医院住院就诊的15例成人MOG-EM患者的资料,15例均按照2020年新发表的《抗髓鞘少突胶质细胞糖蛋白免疫球蛋白G抗体相关疾病诊断和治疗中国专家共识》的标准重核诊断,并参照以下纳入及排除标准[4]。纳入标准:符合以下所有标准,①用全长人MOG作为靶抗原的细胞法检测血清MOG-IgG阳性;②临床有下列表现之一或组合,a.视神经炎,包括慢性复发性炎性视神经病变,b.横贯性脊髓炎(TM),c.脑炎或脑膜脑炎,d.脑干脑炎;③与中枢神经系统脱髓鞘相关的MRI或电生理[孤立性视神经炎(ON)患者的视觉诱发电位(VEP)]检查结果;④排除其他诊断;⑤年龄> 18岁。排除标准:非炎症性的中枢神经系统脱髓鞘病变。复发标准:在急性期治疗后原有临床症状加重或出现新的症状或体征,影像学检查可见新的责任病灶。
二、方法
收集15例患者的临床资料,包括一般资料、临床症状、影像学资料、实验室检查、治疗及预后情况。收集复发患者复发时的临床症状、影像学资料、抗体检测情况。收集患者复查时的临床资料。
三、统计学处理
使用SPSS 20.0对数据进行统计学描述,符合正态分布的计量资料以$\bar{x}\pm s$表示,非正态分布的计量资料以中位数(四分位数间距)表示,计数资料以例数(相对比)表示。
结果
一、一般情况
15例患者中男7例、女8例,起病年龄(39.0 ±14.7)岁,起病至我院就诊时间32.3(32.2)d。起病前有发热3例,均为上呼吸道感染。8例患者出现复发,复发时间间隔为8.4(6.9)个月。15例中7例为视神经炎,5例为脑干脑炎,7例为脑炎,3例为脑膜脑炎,2例为TM。
二、临床特点
本组15例患者中以视力下降最多见,共7例(7/15),4例(4/15)出现行走不稳,5例(5/15)肢体瘫痪或肢体麻木,4例(4/15)出现行走不稳,2例(2/15)记忆力下降,2例(2/15)构音不清,2例(2/15)精神异常,1例(1/15)视物重影,1例(1/15)意识障碍。6例(6/15)伴有头痛,2例(2/15)伴有头晕, 2例(2/15)伴有呕吐。7例以单发临床症状起病,8例以2组或2组以上临床症状同时起病。
8例(8/15)患者出现复发,3例(3/8)复发时表现为行走不稳,2例(2/8)表现为构音不清, 2例(2/8)出现精神症状,1例(1/8)出现视野缺损,1例(1/8)癫痫发作,1例(1/8)出现肢体麻木,1例(1/8)再发头痛。
三、实验室检查
15例患者均进行了脑脊液检查,4例(4/15)脑脊液压力升高,最高达320 mm H2O(1 mm H2O=0.0098 kPa),11例(11/15)压力在正常范围内;8例(8/15)白细胞增多,均< 100×106/L,细胞学分类以淋巴细胞、单核细胞比率升高为主;5例(5/15)蛋白含量升高,但均< 1 g/L。12例行脑脊液寡克隆抗体检测,均阴性。7例患者行ESR检测,6例(6/7)升高。2例患者血清抗链球菌溶血素(ASO)明显升高。1例患者合并抗干燥综合征A抗体(抗SSA抗体)、抗核抗体(ANA)阳性。15例患者血清MOG-IgG阳性,其中11例行脑脊液MOG-IgG检测,5例(5/11)阳性。8例(8/15)患者临床复发。10例复查血清MOG-IgG,5例(5/10)抗体转阴,这5例中4例(4/5)无复发,1例(1/5)临床复发,表现为视力下降加重,影像学检查病灶范围扩大;另5例(5/10)血清MOG-IgG持续阳性,均复发,出现新的临床症状。3例(3/15)血清MOG-IgG阳性合并脑脊液抗谷氨酸受体(NMDA型)抗体阳性,均复发。
四、影像学特点
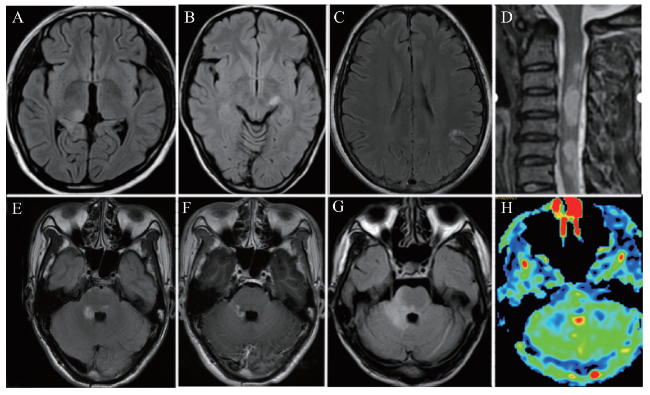
15例患者均行头颅MRI+增强检查,9例行脊髓MRI+增强检查。15例患者中13例(13/15)可见颅内异常病灶,其中2例(2/13)合并脊髓病灶;1例(1/15)头颅MRI正常,脊髓MRI有异常病灶;另1例(1/15)头颅、脊髓MRI均无异常病灶。13例头颅MRI有异常病灶的患者中,病灶累及大脑皮层及皮层下白质8例(8/13),累及桥脑4例(4/13),累及丘脑2例(2/13),累及小脑2例(2/13),累及胼胝体1例(1/13);其中11例(11/13)增强可见病灶强化,多为斑点状、斑片状、线样强化,部分为环形强化,5例(5/13)可见软脑膜强化。9例进行脊髓MRI检查的患者中3例(3/9)可见异常病灶,均累及颈髓,1例累及胸髓。9例患者行动脉自旋标记(ASL)显示7例(7/9)呈低灌注,2例(2/9)呈等灌注。部分患者临床复发,复发后病灶范围扩大。典型病例的MRI检查见图1。
{kind=link}
{kind=link}
五、治疗及预后情况
15例患者均接受静脉应用大剂量甲泼尼龙(500 ~ 1000 mg/d)冲击治疗,3 ~ 5 d后甲泼尼龙序贯递减至小剂量口服,总疗程3个月 ~ 2年;2例急性期联合应用大剂量Ig[400 mg/(kg·d)]冲击治疗,疗程为5 d;1例大剂量甲泼尼龙治疗效果欠佳者加用吗替麦考酚酯(500 mg、2次/日)口服维持治疗。11例患者急性期对大剂量甲泼尼龙冲击治疗及在此基础上联合Ig冲击治疗反应良好,治疗后临床症状明显好转;1例患者对甲泼尼龙治疗反应欠佳,联合免疫抑制剂治疗后有效。所有患者均经临床及电话随访,8例患者临床复发,其中3例反复发作的患者遗留视力下降症状;其余7例患者情况稳定。
讨论
医学界对MOG-EM的认识目前仍处于研究阶段,早期的研究显示其在临床症状与影像学上与MS相似,譬如大多数患者容易复发、15%的成人患者符合MS的诊断标准等,故当时这一疾病被认为属于MS的范围[5]。但后来学者们逐渐发现,一些对MS疗效确切的药物对MOG患者无效或反而加重其病情,且MOG-EM患者在停用激素后容易复发,并对血浆置换、免疫抑制剂等有较好的疗效,这些均与MS明显不同;另外,在AQP4抗体阴性的NMOSD中亦可见MOG-IgG的表达,其临床特点与AQP4抗体阳性的NMOSD亦不相同[6,7,8]。因此,学者们提出MOG-EM的概念,认为其是一种不同于MS和AQP4抗体阳性的NMOSD的独立疾病实体,并在2018年制定其诊断和抗体检测的国际共识[9]。
有关MOG-EM的研究显示其在成年患者中以视神经炎最多见,其次是TM,也可累及大脑半球、脑干、小脑,出现相应的症状[10]。我们通过对15例MOG-EM患者的临床资料进行回顾性分析发现,患者以视神经炎症状起病最多见,其次为肢体瘫痪或肢体麻木、行走不稳、构音不清、视物重影等脑干、小脑症状,与既往报道相符。但我们研究的15例患者中以急性脊髓炎起病的相对较少,且脊髓损害的症状均较轻,其中1例以单个手指麻木为唯一临床表现,脊髓MRI检查发现节段性脊髓损害。这也提示我们在临床上要注意对这一类疾病进行鉴别,对有局灶性神经系统定位体征的患者进行诊断时除需考虑常见的周围性因素外亦需考虑中枢性原因,必要时需进行头颅、脊髓MRI检查,避免漏诊、误诊。
有关MOG-EM和AQP4阳性的NMOSD的影像学研究显示两者病灶的部位不同。MOG-EM患者眼眶MRI常见视神经异常的表现,而脊髓MRI异常病灶在AQP4阳性的NMOSD患者中多见;在累及半球的病灶中,MOG-EM患者以幕上深部白质、皮层灰质及皮层下白质受累多见,脑桥亦较多受累,小脑、中脑、延髓和胼胝体均可受到影响,最不常累及的部位是脑室周围白质、极后区和基底节[11]。本研究中的13例颅内有异常病灶的患者中,8例(8/13)累及大脑皮层及皮层下白质,4例(4/13)累及桥脑,丘脑、小脑、胼胝体均有受累,与上述报道亦相符。9例进行脊髓MRI检查的患者中仅3例(3/9)显示脊髓病灶,均累及颈髓,1例累及胸髓,提示颈髓为成人MOG-EM容易受累的部位。头颅MRI增强检查大部分颅内病灶有强化,部分可见软脑膜强化。9例患者的ASL中7例(7/9)呈低灌注,2例(2/9)呈等灌注。软脑膜强化提示脑膜受累,是脑炎的重要影像学特征,而脑炎在ASL上往往呈高灌注。ASL低灌注是中枢神经系统脱髓鞘病变的典型影像学特点,也是脱髓鞘疾病与其他颅内疾病相鉴别的重要依据。MOG-EM低灌注的影像学特点说明其在病理上仍属于脱髓鞘疾病的范畴,但显著的脑膜强化又提示其有脑炎的特征,影像学上的这些特殊表现也进一步支持MOG-EM是不同于脑炎和MS的独立疾病实体。
MOG-EM脑脊液检查无特异性。我们对15例患者均进行了脑脊液检查,8例白细胞增多,5例蛋白含量升高。11例患者进行脑脊液MOG-IgG检测,5例(5/11)阳性。既往研究显示,MOG-IgG在外周淋巴器官中产生,随后通过血脑屏障渗漏进入中枢神经系统,因此以血清MOG-IgG检测阳性作为确诊MOG-IgG相关疾病的金标准。但也有患者血清及脑脊液MOG-IgG同时阳性,甚至部分患者血清MOG-IgG阴性而脑脊液MOG-IgG阳性。对此,有学者建议同时进行血清及脑脊液MOG-IgG的评估,这样可提高实验室检测敏感性,为MOG/AQP4-IgG血清阴性但高度怀疑为NMOSD或其他相关的炎症性疾病的诊断提供帮助,有助于早期合理治疗。但目前相关的研究仍较少,需要大样本的系统性研究来进一步明确[12]。
与AQP4阳性的NMOSD相比,MOG-EM最开始被认为有较好的预后和较低的临床复发率,但随着对这一疾病认识的深入,学者们发现其同样存在较高的复发率,并会导致严重的视觉和运动功能障碍[13]。有学者对复发患者进行研究,发现这些患者血清MOG-IgG水平往往较高或呈持续阳性, 而单相病程的患者抗体水平随时间的推移会出现滴度的下降或消失[14-15]。一项对50例MOG-EM患者平均33个月的随访研究显示,18例患者出现复发,复发患者血清MOG-IgG滴度在最后随访时仍然呈阳性[16]。本研究中的15例患者均经临床及电话随访,7例恢复良好,无明显后遗症。8例患者临床复发,3例反复发作的患者遗留视力障碍。另有10例患者复查了血清MOG-IgG。复查的10例患者中5例抗体转阴,其中4例无复发;5例抗体呈持续阳性,均复发。这一结果提示持续的血清抗体阳性可能与疾病的复发有一定的关系。但鉴于本研究纳入的病例数较少,尚不能得出相应的结论,今后我们会继续扩大样本量,进行相关的追踪研究。