 PDF(3355 KB)
PDF(3355 KB)
 PDF(3355 KB)
PDF(3355 KB)
 PDF(3355 KB)
PDF(3355 KB)
缝隙连接传递伤害性信号促进小鼠肝缺血再灌注损伤及细胞凋亡
Gap junction-mediated harmful signal transmission contributes to liver injury and cell apoptosis after hepatic ischemia-reperfusion
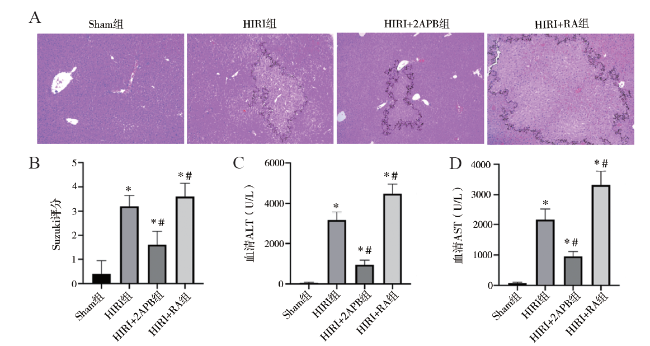
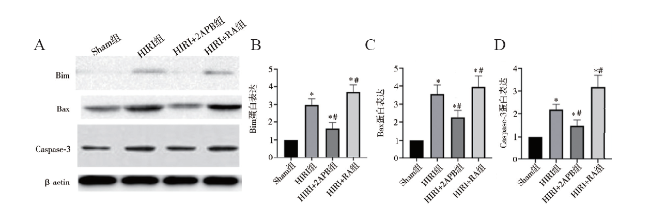
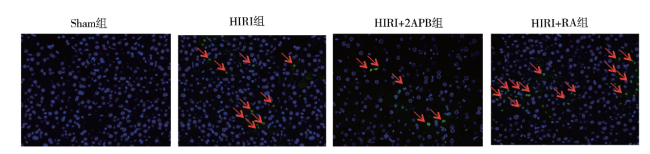
目的 探讨细胞缝隙连接(GJ)信号传递对肝缺血再灌注损伤(HIRI)及细胞凋亡的影响。方法 C57BL/6小鼠建立HIRI模型,随机分为假手术组(Sham组)、HIRI组、HIRI+2-氨基乙基联苯基硼酸酯(2APB)组、HIRI+维甲酸(RA)组。HIRI+2APB组及HIRI+RA组在造模前腹腔注射2APB或RA。采用HE染色观察肝组织病理损伤情况,生化检测血清ALT和AST评估肝功能情况,蛋白免疫印迹法检测凋亡相关蛋白Bim、Bax、Caspase-3的表达,TUNEL法检测肝组织细胞凋亡情况。结果 与HIRI组相比,HIRI+RA组肝损伤及凋亡明显增加,Bim介导的凋亡蛋白表达增强(P均< 0.05);相反,HIRI+2APB组可显著减轻肝损伤及细胞凋亡,并减少Bim介导的凋亡蛋白表达(P均< 0.05)。结论 GJ传递伤害性信号可能在HIRI进展中发挥重要作用。
Objective To evaluate the effect of gap junction (GJ)-mediated signal transmission on liver injury and cell apoptosis after hepatic ischemia reperfusion (HIRI). Methods C57BL/6 mouse models of HIRI were established. All animals were randomly divided into the sham operation group (Sham group), HIRI group, HIRI+2-aminoethyldiphenyl borate (2APB) group and HIRI+retinoic acid (RA) group, respectively. In the HIRI+2APB and HIRI+RA groups, the animals were intraperitoneally injected with 2APB or RA before the establishment of HIRI models. The pathological injury of liver tissues was observed by HE staining. The liver function was evaluated by biochemical detection of serum ALT and AST levels. The expression levels of apoptosis-related proteins, such as Bim, Bax and Caspase-3 in liver tissues was quantitatively measured by Western blot. The cell apoptosis of liver tissues was detected by TUNEL assay. Results Compared with the HIRI group,the liver injury induced by HIRI and the cell apoptosis were aggravated and the expression level of Bim-mediated apoptosis-related protein was remarkably up-regulated in the HIRI+RA group (all P < 0.05). In the HIRI+2APB group, hepatic injury and cell apoptosis were significantly alleviated and the expression level of Bim-mediated apoptosis-related protein was remarkably down-regulated (all P < 0.05). Conclusion The harmful signal transmitted by GJ may play an important role in the development of HIRI-induced liver injury.
肝缺血再灌注损伤 / 凋亡 / 缝隙连接 {{custom_keyword}} /
Hepatic ischemia reperfusion injury / Apoptosis / Gap junction {{custom_keyword}} /
| [1] |
彭娜, 王彦峰, 何维阳, 李明霞, 胡晓燕, 叶啟发. 肝移植供肝缺血再灌注损伤的研究进展. 中华肝胆外科杂志, 2016,22(5):349-351.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [2] |
王冰, 林颖, 刘慧玲, 金海, 汪根树, 吴斌, 陈规划. 己酮可可碱对大鼠肝脏缺血再灌注损伤的影响. 新医学, 2011,42(8):601-604.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [3] |
The BH3-only Bim protein is a major determinant for initiating the intrinsic apoptotic pathway under both physiological and pathophysiological conditions. Tight regulation of its expression and activity at the transcriptional, translational and post-translational levels together with the induction of alternatively spliced isoforms with different pro-apoptotic potential, ensure timely activation of Bim. Under physiological conditions, Bim is essential for shaping immune responses where its absence promotes autoimmunity, while too early Bim induction eliminates cytotoxic T cells prematurely, resulting in chronic inflammation and tumor progression. Enhanced Bim induction in neurons causes neurodegenerative disorders including Alzheimer's, Parkinson's and Huntington's diseases. Moreover, type I diabetes is promoted by genetically predisposed elevation of Bim in beta-cells. On the contrary, cancer cells have developed mechanisms that suppress Bim expression necessary for tumor progression and metastasis. This review focuses on the intricate network regulating Bim activity and its involvement in physiological and pathophysiological processes.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [4] |
Gap junctions, which mediate intercellular communication, are key players in digestive homeostasis. They are also frequently involved in gastrointestinal and liver pathology. This equally holds true for connexin (Cx) hemichannels, the structural precursors of gap junctions, and pannexin (Panx) channels, Cx-like proteins assembled in a hemichannel configuration. Both Cx hemichannels and Panx channels facilitate extracellular communication and drive a number of deteriorative processes, such as cell death and inflammation. Cxs, Panxs, and their channels underlie a wide spectrum of gastrointestinal and liver diseases, including gastritis and peptic ulcer disease, inflammatory intestinal conditions, acute liver failure, cholestasis, hepatitis and steatosis, liver fibrosis and cirrhosis, infectious gastrointestinal pathologies, and gastrointestinal and liver cancer. This could open promising perspectives for the characterization of new targets and biomarkers for therapeutic and diagnostic clinical purposes in the area of gastroenterology and hepatology.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [5] |
Since 1994, a Phase I/II clinical study and radiotherapy have carried out using carbon-ion beams produced with the Heavy Ion Medical Accelerator in Chiba (HIMAC) at National Institute of Radiological Sciences. Now we constructed the new treatment facility for the advanced carbon-ion therapy at HIMAC applying a 3D fast spot scanning system with pencil beams. In the field of fundamental biological studies for high-LET heavy ions, there are some reports regarding bystander effects after exposure to alpha particles derived from 238Pu or He-ion microbeams. However, only limited sets of studies have examined bystander effects after exposure to different ion species heavier than helium, such as carbon ions. We have been investigating bystander cellular responses in both normal human and human tumor cells irradiated with the HIMAC carbon ions. Bystander cell-killing effect was observed in the cells harboring wild-type P53 gene, but not in the P53-mutated cells. Moreover, observed bystander effect was suppressed by treating with a specific inhibitor of gap-junction mediated cell-cell communication. There is clear evidence that the carbon-ion irradiation enables the enhanced cell killing in cells with wild-type P53 gene via gap-junction mediated bystander effect.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [6] |
Drug-induced liver injury (DILI) limits the development and application of many therapeutic compounds and presents major challenges to the pharmaceutical industry and clinical medicine. Acetaminophen-containing compounds are among the most frequently prescribed drugs and are also the most common cause of DILI. Here we describe a pharmacological strategy that targets gap junction communication to prevent amplification of fulminant hepatic failure and acetaminophen-induced hepatotoxicity. We demonstrate that connexin 32 (Cx32), a key hepatic gap junction protein, is an essential mediator of DILI by showing that mice deficient in Cx32 are protected against liver damage, acute inflammation and death caused by liver-toxic drugs. We identify a small-molecule inhibitor of Cx32 that protects against liver failure and death in wild-type mice when co-administered with known hepatotoxic drugs. These findings indicate that gap junction inhibition could provide a pharmaceutical strategy to limit DILI and improve drug safety.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [7] |
Gap junction intercellular communication is involved in ischemia-reperfusion (IR) injury of organs. Connexins are proteins that are critical to the function of gap junctions. To clarify the role of gap junctions in IR injury in liver cells, the function of gap junctions was modulated in an in vitro hypoxia/reoxygenation (H/R) model. BRL-3A rat liver cells, endogenously expressing connexins Cx32 and Cx43, were used to model the process of hepatic IR injury. Suppression of gap junction activity was achieved genetically, using Cx32-specific small interfering RNA (siRNA), or chemically, with pharmacological inhibitors, oleamide, and 18-alpha-GA. BRL-3A cells subjected to H/R exhibited reduced cell survival and pathologies indicative of IR injury. Cx32-specific siRNA, oleamide, and 18-alpha-GA, respectively, decreased gap junction permeability, as assessed by the parachute assay. Pretreatment with Cx32-specific siRNA increased cell survival. Pretreatment with oleamide or 18-alpha-GA did not improve cell survival. Modulating gap junction by Cx32 gene silencing protected BRL-3A liver cells from H/R.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [8] |
BACKGROUND: Nuclear factor-E2-related factor 2 (Nrf2)-mediated antioxidant response is the main protective system of graft-liver against ischemia-reperfusion injury after liver transplantation. Propofol is considered to confer protective effects on different organs; thus, we explored the possibility that whether propofol could attenuate graft-liver injury in a rat autologous orthotopic liver transplantation (AOLT) model and mechanisms were associated with activation of Nrf2 pathway. METHODS: Sprague-Dawley rats were randomly divided into four groups: sham-operated group, saline-treated AOLT group, low-dose propofol intervention group, and high-dose propofol intervention group. Liver injury was determined, and concentration of hydroxyl free radical (*OH), superoxide anion (O2(*-)), and malondialdehyde in the liver tissue were detected. The expression of Keap1, Nrf2, HO-1, and NQO1 were explored by Western blotting, and also the change of Nrf2 and keap1 was assessed by immunofluorescence. RESULTS: Compared with sham group, pathologic damage of graft-livers was in a time-dependent manner, accompanied with the increased level of oxidative stress in the AOLT group, and nuclear Nrf2 expression and its downstream antioxidant enzyme, HO-1 and NQO1, were also increased in this group. However, in propofol pretreatment groups especially in the high-dose group, the pathologic score was significantly decreased, accompanied with a lower level of *OH, O2(*-), and malondialdehyde than that of the AOLT group. The change of oxidative stress might be related to the Nrf2 pathway, evidenced as the elevation of protein expression level of NQO1, HO-1, and nuclear Nrf2. CONCLUSIONS: Protective effects of propofol against liver transplantation-induced graft-liver injury may be related with Keap1-Nrf2 signal pathway activation.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [9] |
Aim. To investigate whether hyperglycemia will aggravate hepatic ischemia reperfusion injury (HIRI) and the underlying mechanisms. Methods. Control and streptozotocin-induced diabetic Sprague-Dawley rats were subjected to partial hepatic ischemia reperfusion. Liver histology, transferase, inflammatory cytokines, and oxidative stress were assessed accordingly. Similarly, BRL-3A hepatocytes were subjected to hypoxia/reoxygenation (H/R) after high (25 mM) or low (5.5 mM) glucose culture. Cell viability, reactive oxygen species (ROS), and activation of nuclear factor-erythroid 2-related factor 2 (Nrf2) and nuclear factor of kappa light polypeptide gene enhancer in B-cells (NF-kappaB) were determined. Results. Compared with control, diabetic rats presented more severe hepatic injury and increased hepatic inflammatory cytokines and oxidative stress. HIRI in diabetic rats could be ameliorated by pretreatment of N-acetyl-L-cysteine (NAC) or apocynin. Excessive ROS generation and consequent Nrf2 and NF-kappaB translocation were determined after high glucose exposure. NF-kappaB translocation and its downstream cytokines were further increased in high glucose cultured group after H/R. While proper regulation of Nrf2 to its downstream antioxidases was observed in low glucose cultured group, no further induction of Nrf2 pathway by H/R after high glucose culture was identified. Conclusion. Hyperglycemia aggravates HIRI, which might be attributed to chronic oxidative stress and inflammation and potential malfunction of antioxidative system.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [10] |
黑子清. 肝移植围术期器官损伤机制及器官保护策略研究进展. 中山大学学报(医学版), 2019,40(4):488-492.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [11] |
Hepatic ischemia/reperfusion (I/R) injury is a common complication in the clinical setting. Our previous study has shown that connexin 32 (Cx32) plays a major role in renal I/R injury; however, the role of Cx32 in hepatic I/R injury remains unknown. Liver tissue and serum samples from patients undergoing orthotopic liver transplantation (OLT) were used to evaluate the function of Cx32 in OLT post-reperfusion injury. Then, partial hepatic ischemia was established in global Cx32 knockout mice and wild-type mice followed by reperfusion. Hepatic injury markers were examined. Cx32 small interfering RNA and the p53 inhibitor, pifithrin-alpha, tenovin-1 were used to examine the relationship between Cx32 and the p53/puma pathways in the BRL-3A and murine primary hepatocytes hypoxia/reoxygenation (H/R) model. Corresponding to liver damage, Cx32 was significantly induced both during OLT in human patients and partial hepatic I/R in mice. Cx32 KO mice exhibited less liver injury than controls. Cx32 deficiency significantly suppressed the p53/puma pathways and hepatocyte apoptosis. Similar results were observed in the BRL-3A and murine primary hepatocytes H/R model. Propofol protected against OLT post-reperfusion injury and hepatocyte apoptosis by inhibiting Cx32. In conclusion Cx32 is a novel regulator of hepatic I/R injury through the modulation of hepatocyte apoptosis and damage, largely via the p53/puma signaling pathway.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [12] |
Alcohol-related liver disease (ALD) accounts for the majority of cirrhosis and liver-related deaths worldwide. Activation of IFN-regulatory factor (IRF3) initiates alcohol-induced hepatocyte apoptosis, which fuels a robust secondary inflammatory response that drives ALD. The dominant molecular mechanism by which alcohol activates IRF3 and the pathways that amplify inflammatory signals in ALD remains unknown. Here we show that cytoplasmic sensor cyclic guanosine monophosphate-adenosine monophosphate (AMP) synthase (cGAS) drives IRF3 activation in both alcohol-injured hepatocytes and the neighboring parenchyma via a gap junction intercellular communication pathway. Hepatic RNA-seq analysis of patients with a wide spectrum of ALD revealed that expression of the cGAS-IRF3 pathway correlated positively with disease severity. Alcohol-fed mice demonstrated increased hepatic expression of the cGAS-IRF3 pathway. Mice genetically deficient in cGAS and IRF3 were protected against ALD. Ablation of cGAS in hepatocytes only phenocopied this hepatoprotection, highlighting the critical role of hepatocytes in fueling the cGAS-IRF3 response to alcohol. We identified connexin 32 (Cx32), the predominant hepatic gap junction, as a critical regulator of spreading cGAS-driven IRF3 activation through the liver parenchyma. Disruption of Cx32 in ALD impaired IRF3-stimulated gene expression, resulting in decreased hepatic injury despite an increase in hepatic steatosis. Taken together, these results identify cGAS and Cx32 as key factors in ALD pathogenesis and as potential therapeutic targets for hepatoprotection.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [13] |
2-Aminoethoxydiphenyl borate (2-APB), an inositol 1,4,5-triphosphate receptor modulator, inhibits capacitive current transients measured in normal rat kidney and human embryonic kidney 293 cells, an indication of blocking gap junction channels between these cells. Here, we used the dual whole-cell patch-clamp method to study the actions of 2-APB on gap junction channels formed by selected connexins expressed in a communication-deficient neuroblastoma cell line (N2A). 2-APB dose-dependently and reversibly blocked junctional currents of connexin (Cx) 50 gap junction channels. The concentration-inhibition curve of 2-APB on the junctional current indicated an IC(50) of 3.7 microM, lower than that of most gap junction inhibitors. At a concentration of 20 microM, 2-APB also significantly blocked junctional conductance in cell pairs coupled by Cx26, Cx30, Cx36, Cx40, and Cx45 but did not appreciably affect coupling in cell pairs expressing Cx32, Cx43, and Cx46. Although concentration inhibition curves of 2-APB on Cx36 channels were similar to Cx50 (Cx36; IC(50), 3.0 microM), IC(50) values were higher for Cx43 (51.6 microM), Cx45 (18.1 microM), and Cx46 (29.4 microM). The blocking action of 2-APB did not substantially alter transjunctional voltage-dependent gating of Cx50 gap junction channels, and recordings from poorly coupled pairs of Cx50-transfected N2A cells indicated that 2-APB reduced gap junction channel open probability without changing the main state single-channel conductance. The differential efficacy of block by 2-APB of gap junction channels formed by different connexins may provide a useful tool that could be exploited in gap junction research to selectively block certain gap junction channel subtypes.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [14] |
It has been widely accepted that mitochondria-dependent apoptosis initiates when select BH3-only proteins (BID, BIM, etc.) directly engage and allosterically activate effector proteins BAX/BAK. Here, through reconstitution of cells lacking all eight pro-apoptotic BH3-only proteins, we demonstrate that all BH3-only proteins primarily target the anti-apoptotic BCL-2 proteins BCL-xL/MCL-1, whose simultaneous suppression enables membrane-mediated spontaneous activation of BAX/BAK. BH3-only proteins' apoptotic activities correlate with affinities for BCL-xL/MCL-1 instead of abilities to directly activate BAX/BAK. Further, BID and BIM do not distinguish BAX from BAK or accelerate BAX/BAK activation following inactivation of BCL-xL/MCL-1. Remarkably, death ligand-induced apoptosis in cells lacking BH3-only proteins and MCL-1 is fully restored by BID mutants capable of neutralizing BCL-xL, but not direct activation of BAX/BAK. Taken together, our findings provide a
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [15] |
The pro-apoptotic Bcl-2-family protein Bim belongs to the BH3-only proteins known as initiators of apoptosis. Recent data show that Bim is constitutively inserted in the outer mitochondrial membrane via a C-terminal transmembrane anchor from where it can activate the effector of cytochrome c-release, Bax. To identify regulators of Bim-activity, we conducted a search for proteins interacting with Bim at mitochondria. We found an interaction of Bim with Tom70, Tom20 and more weakly with Tom40, all components of the Translocase of the Outer Membrane (TOM). In vitro import assays performed on tryptically digested yeast mitochondria showed reduced Bim insertion into the outer mitochondrial membrane (OMM) indicating that protein receptors may be involved in the import process. However, RNAi against components of TOM (Tom40, Tom70, Tom22 or Tom20) by siRNA, individually or in combination, did not consistently change the amount of Bim on HeLa mitochondria, either at steady state or upon de novo-induction. In support of this, the individual or combined knock-downs of TOM receptors also failed to alter the susceptibility of HeLa cells to Bim-induced apoptosis. In isolated yeast mitochondria, lack of Tom70 or the TOM-components Tom20 or Tom22 alone did not affect the import of Bim into the outer mitochondrial membrane. In yeast, expression of Bim can sensitize the cells to Bax-dependent killing. This sensitization was unaffected by the absence of Tom70 or by an experimental reduction in Tom40. Although thus the physiological role of the Bim-TOM-interaction remains unclear, TOM complex components do not seem to be essential for Bim insertion into the OMM. Nevertheless, this association should be noted and considered when the regulation of Bim in other cells and situations is investigated.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [16] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [17] |
BH3-only proteins constitute major proportion of pro-apoptotic members of B-cell lymphoma 2 (Bcl-2) family of apoptotic regulatory proteins and participate in embryonic development, tissue homeostasis and immunity. Absence of BH3-only proteins contributes to autoimmune disorders and tumorigenesis. Bim (Bcl-2 Interacting Mediator of cell death), most important member of BH3-only proteins, shares a BH3-only domain (9-16 aa) among 4 domains (BH1-BH4) of Bcl-2 family proteins and highly pro-apoptotic in nature. Bim initiates the intrinsic apoptotic pathway under both physiological and patho-physiological conditions. Reduction in Bim expression was found to be associated with tumor promotion and autoimmunity, while overexpression inhibited tumor growth and drug resistance as cancer cells suppress Bim expression and stability. Apart from its role in normal homeostasis, Bim has emerged as a central player in regulation of tumorigenesis, therefore gaining attention as a plausible target for chemotherapy. Regulation of Bim expression and stability is complicated and regulated at multiple levels viz. transcriptional, post-transcriptional, post-translational (preferably by phosphorylation and ubiquitination), epigenetic (by promoter acetylation or methylation) including miRNAs. Furthermore, control over Bim expression and stability may be exploited to enhance chemotherapeutic efficacy, overcome drug resistance and select anticancer drug regimen as various chemotherapeutic agents exploit Bim as an executioner of cell death. Owing to its potent anti-tumorigenic activity many BH3 mimetics e.g. ABT-737, ABT-263, obatoclax, AT-101and A-1210477 have been developed and entered in clinical trials. It is more likely that in near future strategies commanding Bim expression and stability ultimately lead to Bim based therapeutic regimen for cancer treatment.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [18] |
Agents that inhibit bromodomain and extra-terminal domain (BET) protein have been actively tested in the clinic as potential anticancer drugs. Proteasome inhibitors such as carfilzomib (CFZ) are FDA-approved for the treatment of patients with advanced multiple myeloma and have been tested against other cancers. The current study focuses on the combination of a BET inhibitor (e.g., JQ1) and a proteasome inhibitor (e.g., CFZ) as a novel cancer therapeutic strategy and the underlying mechanisms. The tested combination (JQ1 with CFZ) synergistically decreased cell survival and enhanced apoptosis in vitro and inhibited tumor growth in vivo. The dramatic induction of apoptosis was accompanied by enhanced elevation of Bim and ER stress. Bim knockout significantly attenuated apoptosis induced by the combination, suggesting a critical role of Bim induction in mediating the enhanced induction of apoptosis by BET and proteasome co-inhibition. The combination significantly increased Bim mRNA levels with limited effect on Bim protein stability, suggesting a primary transcriptional regulation of enhanced Bim expression. Our findings warrant further investigation of this combinatorial strategy as an effective regimen against cancer in the clinic.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| {{custom_ref.label}} |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
PDF(3355 KB)
/
| 〈 |
|
〉 |




{kind=link}
{kind=link}
{kind=link}