


溃疡性结肠炎(UC)是一种慢性非特异性炎症性肠病(IBD),主要累及结直肠黏膜层及黏膜下层。血性腹泻、腹痛是UC最常见的临床症状。目前病因尚不明确,研究表明其与环境、遗传易感性、肠道菌群失调和免疫反应紊乱等因素相关[1]。自噬是真核细胞的一个基本生物过程,自噬通过对蛋白质和细胞器的分解代谢,在能量稳态中起重要作用,主要分为巨自噬、微自噬和分子伴侣介导的自噬等[2]。巨自噬是指内质网或高尔基体来源的隔膜在待降解物质周围形成自噬小体,运输至溶酶体内进行降解的过程。全基因组关联研究确定IBD的多个易感基因,大量的研究表明自噬在维持肠道内稳态中发挥着重要作用,可能与IBD的发病机制相关[3]。β-抑制蛋白2 (β-arrestin2)是Arrestins家族中的一员,是广泛存在于细胞膜和细胞质内的支架蛋白[4]。β-arrestin2不仅介导G蛋白偶联受体(GPCR)的脱敏、内化和信号转导;同时还通过参与胰岛素生长因子1(IGF-1)、核因子-κB (NF-κB)、p53、Wnt、PI3K/Akt等多条信号通路,进而调节细胞的趋化、凋亡、自噬等功能,在许多生理和病理过程中起着重要作用[5]。笔者团队既往的研究发现β-arrestin2可以促进葡聚糖硫酸钠(DSS)诱导的结肠炎黏膜的细胞凋亡,然而在结肠炎中,β-arrestin2是否对自噬存在调控作用尚不清楚[6]。本研究通过建立β-arrestin2野生型(WT)和β-arrestin2基因敲除型(KO)小鼠急性UC模型以及干扰β-arrestin2在HCoEpiC细胞中的表达,探讨β-arrestin2在小鼠UC中对自噬水平的调控。
材料与方法
一、材料
1. 实验动物
SPF级别的C57BL/6J小鼠20只,雌雄各半,8周龄,体质量20~25 g,购自广东药康生物科技有限公司。β-arrestin2 KO小鼠于中山大学附属第三医院疫苗研究所动物中心进行繁育、饲养并用于实验。本动物实验伦理经中山大学实验动物管理与使用委员会审批通过(F2-09-0901)。
2. 实验细胞系
人源的正常结肠上皮细胞系HCoEpiC细胞由中山大学附属第三医院消化内科实验室冻存保种。
3. 主要试剂与抗体
DSS(MP Biomedicals,美国)、高糖DMEM培养基(Gibco,美国)、胎牛血清(Gibco,美国)、β-arrestin2 siRNA(吉玛基因,中国)、β-actin抗体(Santa Cruz,美国)、微管相关蛋白1轻链3β(LC3B)抗体(Abcam,美国)、 Beclin1抗体(CST,美国)、p-Akt(CST,美国)、β-arrestin2抗体(Proteintech,中国)、Earle's平衡盐溶液(EBSS,Gibco,美国)。
4. 主要仪器
二氧化碳培养箱(Thermo Scientific,美国)、SDS-PAGE电泳槽(BIO-RAD,美国)、半干转膜仪(BIO-RAD,美国)、PCR仪(BIO-RAD,美国)。
二、方法
1. 小鼠急性UC模型的建立
选取8周大的C57BL/6J小鼠,随机分为β-arrestin2 WT对照组、β-arrestin2 WT实验组、β-arrestin2 KO对照组和β-arrestin2 KO实验组,每组10只。WT对照组和KO对照组小鼠给予无菌的ddH2O自由饮用,WT实验组和KO实验组小鼠自由饮用含3%DSS的无菌ddH2O 7 d,诱导小鼠急性UC。
2. 疾病活动指数评分
从造模的第1日起,每日观察小鼠的体质量、粪便性状和粪便隐血情况,计算小鼠疾病活动指数。具体评分标准如下:体质量正常为0分,体质量下降1%~5%为1分,下降5%~10%为2分,下降10%~20%为3分,下降大于20%为4分;大便成形为0分,呈糊状或半成形为1分,稀便为4分;粪便潜血阴性为0分,大便潜血阳性为2分,肉眼血便为4分。
3. 小鼠结肠标本的收集
造模结束后,对小鼠进行腹腔注射10%水合氯醛,深度麻醉后行颈椎脱臼处死。剖开腹部暴露腹腔,取出全结肠,放置冰盒上。纵行剖开小鼠结肠,用预冷的生理盐水冲洗结肠内容物。部分结肠组织进行固定后续包埋,部分结肠组织刮取肠黏膜后置于-80℃冰箱中保存,后续提取蛋白质。
4. HE染色
将制备好的蜡块切片,进行脱蜡和梯度水化处理后,滴加苏木素染液30 s,流水冲洗后置于磷酸盐缓冲液(PBS)中反蓝。随后用伊红染液染色1 min,流水冲洗后显微镜下观察染色情况,封片,拍照。
5. 免疫组织化学染色
将制备好的蜡块切片,进行脱蜡和梯度水化处理后,在3%双氧水中孵育10 min,然后进行抗原修复,用5%的牛血清白蛋白封闭30 min。一抗稀释液4℃孵育过夜,PBS清洗,对应的二抗稀释液37℃孵育2 h。PBS冲洗后DAB显色,蒸馏水终止反应。苏木素染液染色后,封片,拍照。
6. 细胞培养
将HCoEpic细胞用完全培养基,置于5% CO2、37℃培养箱中进行培养,观察细胞的生长状况。待细胞处于对数增长期时,胰酶消化,用培养基重悬细胞,按1∶4的比例传代。
7. siRNA沉默β-arrestin2基因的表达
当细胞生长到50%的密度时,根据siRNA说明书进行瞬时转染,沉默β-arrestin2基因的表达。siRNA干扰效率通过实时荧光定量PCR(RT-PCR)和蛋白免疫印迹进行检测。将消化重悬的细胞铺于12孔板中,使24 h后达到50%,用siRNA沉默β-arrestin2的表达。EBSS处理细胞2 h。
8. 细胞RNA提取及RT-PCR检测mRNA表达
根据试剂盒说明书提取细胞中的总RNA,测定提取的RNA样品的纯度及浓度,逆转录后测定目的基因的mRNA表达水平。目的基因引物序列见表1。
表1 目的基因引物序列 |
| 基因 | 来源 | 上游引物 | 下游引物 |
|---|---|---|---|
| β-actin | 人 | 5′-GGCTGTATTCCCCTCCATCG-3′ | 5′-CCAGTTGGTAACAATGCCATGT-3′ |
| β-arrestin2 | 人 | 5′-GATGTGCTGGGCTTGTCCTTC-3′ | 5′-ATGGAAGATTCTGGGGTATGGT-3′ |
9. 结肠黏膜和细胞总蛋白提取及蛋白免疫印迹
根据总蛋白提取裂解液说明书对结肠黏膜细胞总蛋白进行提取和定量。取蛋白样品进行电泳,转膜结束后,封闭2 h。用磷酸盐吐温缓冲液洗涤后,一抗4℃孵育过夜。磷酸盐吐温缓冲液洗涤后,对应的二抗室温孵育2 h,化学发光法显像,采用Image J图像分析软件进行灰度分析。
三、统计学处理
所得数据应用SPSS 21.0进行统计学分析。正态分布数据均以 $\bar{x} \pm s$ 表示,2组间比较采用两独立样本t检验。P < 0.05为差异具有统计学意义。采用GraphPad Prism 8.0软件进行作图。
结果
一、小鼠急性UC的结肠黏膜自噬水平上调
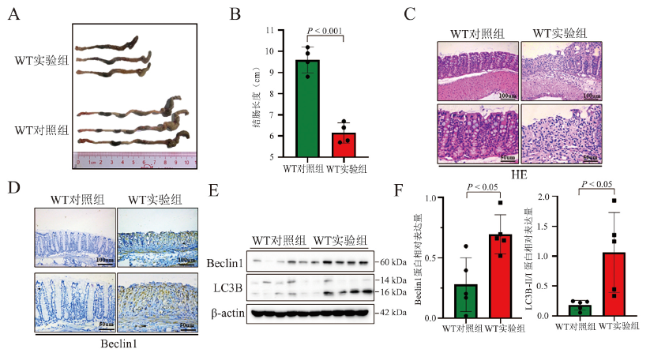
二、β-arrestin2的缺失减轻小鼠急性UC的炎症反应
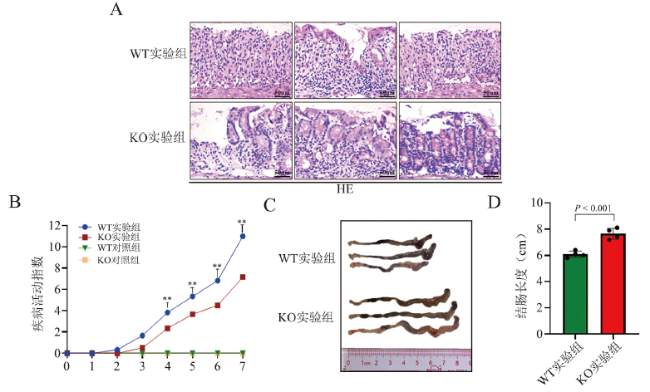
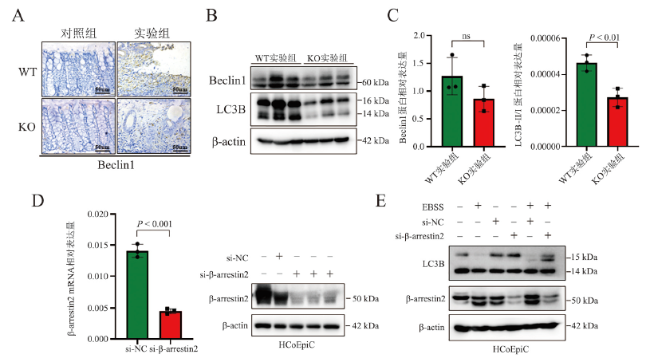
三、β-arrestin2的缺失下调小鼠UC结肠黏膜和HCoEpiC细胞中的自噬水平
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图3 各组小鼠急性UC结肠黏膜和HCoEpiC细胞中的自噬水平比较注:A为DSS造模前后,4组小鼠结肠黏膜Beclin1免疫组织化学染色(标尺50 μm);B为DSS造模后,实验组小鼠结肠黏膜LC3B、Beclin1蛋白免疫印迹电泳图;C为DSS造模后,实验组小鼠结肠黏膜LC3B-Ⅱ/Ⅰ、Beclin1蛋白相对表达量比较,ns为P > 0.05;D为在HCoEpiC细胞中,siRNA沉默β-arrestin2的表达后,β-arrestin2 mRNA水平相对表达量和蛋白免疫印迹电泳图;E为siRNA沉默HCoEpiC细胞中β-arrestin2表达后,采用EBSS处理细胞2 h,LC3B 蛋白免疫印迹电泳图。 |
讨论
UC是一种累及结直肠的慢性非特异性炎症疾病,与多种致病因素相关,被认为与环境因素、遗传易感性、肠道微生物失调以及免疫功能紊乱有关[7-8]。β-arrestins几乎在所有组织细胞上表达,主要分为β-arrestin1和β-arrestin2,其中β-arrestin1存在于细胞质和细胞核中,而β-arrestin2仅存在于细胞质中[4]。作为衔接蛋白,β-arrestin2不仅介导GPCR的内吞和脱敏,是GPCR信号转导的负向调节因子;同时又参与多种信号通路的转导,调节细胞的增殖、凋亡、自噬、迁移、炎症等功能[5]。大量研究表明β-arrestin2可能通过多种途径调节炎症反应。β-arrestin2可以通过与IκBα结合从而阻止NF-κB降解,或与TRAF6结合从而阻止TRAF6寡聚化和自泛素化,从而抑制NF-κB的激活[9-10]。同时,β-arrestin2可以抑制自然杀伤细胞的毒性作用,参与调节T淋巴细胞的活化等[11⇓-13]。β-arrestin2在肠道炎症中起保护作用还是损伤作用仍存在争议。已有研究表明,β-arrestin2可以通过调节Th1和Th17细胞的比例从而抑制肠道炎症的进展[13]。而β-arrestin2又介导肥大细胞对肠道通透性的调节,削弱结肠细胞间的紧密连接,增加细胞间的通透性[14]。本实验在β-arrestin2 WT小鼠和β-arrestin2 KO小鼠上建立了急性UC模型,通过HE染色、疾病活动指数、结肠长度对比等分析方法,再次验证了β-arrestin2的缺失可以减轻小鼠急性UC的炎症反应。
同时,既往研究报道,全基因组关联性研究证实了与IBD相关的多个基因位点的单核苷酸多态性如ATG16L、NOD2、LRRK2、IRGM等分别调控IBD中的自噬发生[3⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓, 16]。研究表明,具有ATG16L1的单核苷酸多态性的人类细胞自噬受损,对病原体的清除能力减弱,潘氏细胞和杯状细胞功能紊乱[17]。目前已有大量的研究报道自噬在肠道固有免疫和适应性免疫中发挥着重要的作用。在固有免疫中,自噬与模式识别受体相互作用,如Toll样受体、NOD样受体等,识别并消除侵入细胞的病原体。同时在肠道中,内源性和外源性微生物可通过自噬进行抗原呈递,并对共生物抗原耐受性的产生具有重要作用[18]。许多自噬相关蛋白(如Beclin1、Atg3、Atg5和Atg7)对T淋巴细胞的生存、发育和成熟是必要的[19-20]。干扰自噬导致肠道内固有免疫和适应性免疫受损,自噬缺陷可影响细胞抗原肽的呈递、损害细胞内细菌清除能力等[16⇓-18]。然而,自噬是把双刃剑,在炎症中具有双向调节作用,适当的自噬可以降解受损或衰老的细胞器以及错误折叠的蛋白质,而过度的自噬会引起细胞自噬性死亡。有研究报道,过度的自噬会损害上皮细胞的紧密连接,抑制过度的自噬,从而恢复ZO-1和E-cadherin的表达[21]。为此,我们在实验中先探究了小鼠急性UC模型中自噬水平的变化,通过蛋白免疫印迹、免疫组织化学等检测方法,发现诱导小鼠急性UC后,自噬相关蛋白的表达水平上调。而β-arrestin2的缺失可以改善小鼠结肠黏膜的炎症,于是我们进一步探究β-arrestin2是否可以通过调控自噬来调节肠道的炎症。因此我们分析了β-arrestin2 WT小鼠和β-arrestin2 KO小鼠在急性UC模型中自噬水平的变化,结果发现β-arrestin2的缺失可以抑制自噬的发生,进一步在人源性的结肠上皮细胞HCoEpic中进行验证。通过饥饿诱导细胞自噬的发生,发现沉默β-arrestin2表达的细胞中,自噬相关蛋白表达水平下降,自噬水平受到抑制。
综上所述,β-arrestin2的缺失抑制自噬的发生,缓解肠道的炎症反应,为UC的治疗提供新的理论依据。