


人类的肠道中含有超过100万亿个微生物,它们影响着宿主的营养、代谢、生理和免疫功能[1]。肠道微生物包括细菌、真菌、古生菌、原生动物和病毒等,主要以细菌为主[2]。多项研究表明,慢性肾脏病(CKD)患者肠道菌群的种类和数量都较健康人群发生了变化,导致致病菌的增多或肠道代谢产物的蓄积,这些都会进一步影响疾病的进程和预后[3,4]。当CKD发展到终末期肾脏病(ESRD)时,最常见的治疗为血液透析和腹膜透析,因血液透析的患者会以动静脉瘘/移植物或静脉导管的形式存在血管通路以及严格的饮食限制,而腹膜透析存在长期的腹膜导管和不断流入的含有微生物营养素的腹膜透析液,这些都可能进一步影响人类肠道菌群的组成及功能[5]。目前关于血液透析及腹膜透析对肠道菌群影响的研究较少,相关研究主要集中于将ESRD患者或替代治疗的患者肠道菌群与健康对照者比较,没有与未接受血液透析或腹膜透析治疗的ESRD患者相比,因为肠道菌群的改变可能是替代治疗或CKD本身(或两者兼有)的结果。针对这一问题,进一步确认血液透析和腹膜透析对ESRD患者肠道菌群的影响十分必要。为此,本研究选择了健康志愿者、未透析的ESRD患者和行腹膜透析或血液透析的ESRD患者,使用16S rDNA测序分析其粪便样品中肠道微生物群的组成和功能,现报告如下。
对象与方法
一、研究对象
选择2018年8月至2019年2月就诊于我院的健康体检者或ESRD患者。ESRD的诊断标准:利用CKD流行病学公式计算肾小球滤过率,肾小球滤过率 < 15 ml/(min·1.73 m2)。排除标准:腹泻者,不能自行进食或使用肠内、外营养干预者,胃肠道肿瘤、胆道炎症、炎症性肠病等消化道疾病者,近3个月有局部炎症、全身性感染者,近3个月服用抗生素、免疫抑制剂、糖皮质激素、益生菌等药物者。共纳入109例研究对象,分为4组:肾功能正常的健康志愿者19名(健康对照组)、未透析的ESRD患者38例(未透析组)、持续规律腹膜透析6个月以上的ESRD患者25例(PD组)、持续规律血液透析6个月以上的ESRD患者27例(HD组)。本研究操作程序遵循中山大学附属第三医院医学伦理委员会制订的伦理学标准,所有操作均征得研究对象或其亲属的知情同意。
二、研究方法
1. 样品采集
用无菌棉签取中段成型大便放入无菌大便盒,做好标记,迅速放入超低温冰箱中储存。
2. 肠道菌群的检测
采用16S rDNA测序分析,通过粪便DNA基因组提取试剂盒(美国Qiagen)提取粪便样品中的 DNA,特异性扩增样品中的微生物 16S rDNA的高变区,采用高通量测序平台 Hiseq 或 Miseq 对高变区序列进行测序,然后通过生物信息学方法进行序列分析和物种注释,了解样品的群落组成情况。
3. 数据分析
使用Illumina Hiseq 或 Miseq 测序平台获得的原始测序序列,对原始数据进行剪切过滤,获得有效数据;通过测序序列之间的重叠关系将序列贴上标签,在给定的相似度下将标签聚类成可操作分类单元(OTU),选取 OTU 的代表性序列,与数据库进行比对获得物种注释,从而得到每个样品的群落组成信息。肠道菌群的多样性可用α多样性和β多样性进行分析,α多样性中的Observed species指数和Chao1指数反映样品中群落的丰度,即仅考虑群落中物种的数量,而不考虑群落中每个物种的丰度情况。α多样性中的Simpson指数同时考虑了物种组成的丰度和均匀度2个因素,Simpson指数越高表示肠道菌群分布越不均匀[6]。基于OTU代表序列构建系统发育树,结合物种丰度信息,通过主坐标分析(PCoA),进一步比较不同样品和分组的群落结构差异。通过 LDA 分析(线性回归分析)在各组之间进行两两比较,当中有显著作用的微生物类群获得LDA 分值(筛选条件LDA分值> 2.0,P < 0.05),展现组中丰度有显著差异的菌。β多样性用Unifrac距离矩阵比较生物的多样性。Unifrac距离矩阵分为Weighted(加权)和Unweighted(非加权)2种方法,对不同样品间的微生物群落构成进行比较。其中加权计算方法仅考虑物种有无的变化,对稀有物种比较敏感,加权计算方法同时考虑物种有无和物种丰度的变化,对丰度较高的物种更加敏感。同时记录距患者采集大便标本当日最近1次的实验室相关检查数据。
三、统计学处理
通过 LEfSe对分组样品的物种组成和群落结构差异进行统计检验,区别2个或2个以上生物条件(或者是类群)。该算法强调的是统计意义和生物相关性。使用SPSS 23.0进行数据处理,正态分布的计量资料以$\bar{x}±s$表示,多组间比较采用单因素方差分析,组间两两比较采用LSD-t检验;非正态分布的计量资料以中位数(上、下四分位数)表示,2组间比较采用Mann-Whitney U检验,多组间比较Kruskal-Wallis检验;计数资料以百分率表示,组间比较采用$\chi$2检验。总体比较P < 0.05为差异有统计学意义,非正态分布的计量资料4组间两两比较用Bonferroni 法校正检验水准,P < 0.05/6 = 0.008为差异有统计学意义。
结 果
一、健康对照组、未透析组、PD组、HD组的临床资料比较
健康对照组、未透析组、PD组、HD组的性别、年龄、BMI、血脂比较差异均无统计学意义(P均> 0.05),4组的血尿素氮、血肌酐、肾小球滤过率、血红蛋白、血清白蛋白、血钾、血钙、血磷比较差异有统计学意义(P均< 0.05)。未透析组、PD组、HD组的糖尿病、高血压病患者比例比较差异无统计学意义(P均> 0.05),PD组与HD组的透析时间及透析充分性比较差异均无统计学意义(P均> 0.05),见表1。
表1 健康对照组、未透析组、PD组、HD组的临床资料比较 |
| 项 目 | 健康对照组(19例) | 未透析组(38例) | PD组(25例) | HD组(27例) | $\chi$2/t/Z/ F/H值 | P值 |
|---|---|---|---|---|---|---|
| 男性[例(%)] | 9(47) | 18(47) | 16(64) | 15(56) | 2.010 | 0.581 |
| 年龄(岁) | 46.5±6.7 | 49.0±13.0 | 55.0±10.5 | 52.9±14.5 | 7.639 | 0.054 |
| BMI(kg/m2) | 23.1±1.1 | 22.6±3.1 | 23.6±3.5 | 21.8±3.0 | 7.401 | 0.060 |
| 糖尿病[例(%)] | - | 13(34) | 9(36) | 10(37) | 0.058 | 1.000 |
| 高血压病[例(%)] | - | 33(87) | 25(100) | 24(93) | 3.459 | 0.225 |
| 总胆固醇(mmol/L) | 4.57±0.46 | 4.26±1.09 | 4.37±0.93 | 3.85±0.94 | 1.605 | 0.194 |
| 甘油三酯(mmol/L) | 1.41(0.90,1.99) | 1.17(0.91,1.69) | 0.87(0.67,1.83) | 1.20(0.79,1.68) | 1.773 | 0.621 |
| HDL-C(mmol/L) | 1.08±0.15 | 1.11±0.33 | 1.01±0.28 | 1.04±0.27 | 0.530 | 0.663 |
| LDL-C(mmol/L) | 2.92±0.82 | 2.58±0.92 | 2.68±0.85 | 2.26±0.76 | 1.546 | 0.208 |
| 血清铁(μmol/L) | 12.4(5.6,19.2) | 11.2(6.1,16.3) | 9.8(5.8,13.9) | 9.3(7.4,11.6) | 0.539 | |
| 血红蛋白(g/L) | 132±21 | 84±20 | 95±17 | 105±18 | 20.707 | < 0.001 |
| 血清白蛋白(g/L) | 44±4 | 36±4 | 32±3 | 40±3 | 24.912 | < 0.001 |
| 透析时间(年) | - | - | 3.0(1.5,3.3) | 3.0(2.0,6.0) | 1.106 | 0.269 |
| 透析充分性(Kt/V) | - | - | 1.8(1.6,2.1) | 1.6(1.5,1.9) | -1.906 | 0.057 |
| 血尿素氮(mmol/L) | 4.5±1.6 | 26.7±10.4 | 17.9±5.8 | 24.6±5.9 | 40.724 | < 0.001 |
| 血肌酐(μmol/L) | 70±19 | 865±341 | 1 001±236 | 1 085±270 | 34.182 | < 0.001 |
| 肾小球滤过率[ml/(min·1.73 m2)] | 101(90,113) | 6(3,8) | 4(4,5) | 4(3,4) | 28.225 | < 0.001 |
| 血钙(mmol/L) | 2.31±0.14 | 2.12±0.25 | 2.26±0.24 | 2.27±0.25 | 2.795 | 0.045 |
| 血磷(mmol/L) | 1.04±0.34 | 1.70±0.41 | 1.42±0.40 | 1.86±0.53 | 6.244 | 0.001 |
| 血钾(mmol/L) | 4.07±0.36 | 4.29±0.74 | 3.73±0.62 | 4.80±0.71 | 9.887 | < 0.001 |
二、肠道菌群多样性的改变
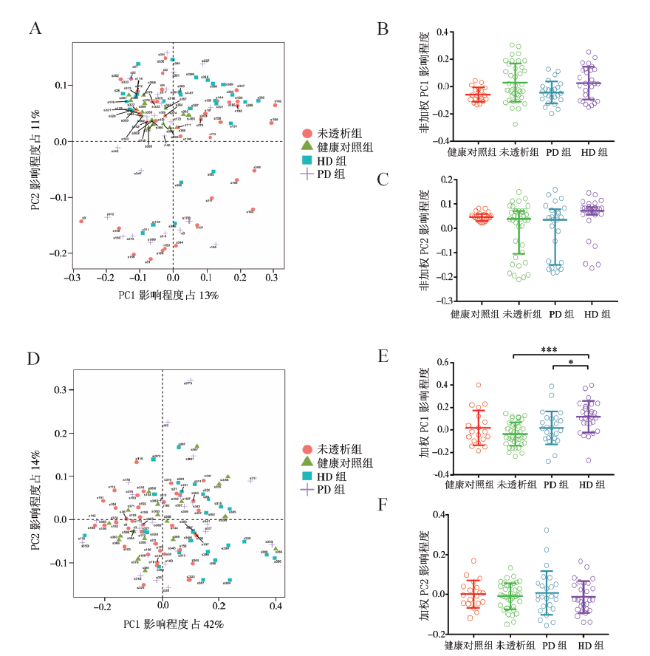
健康对照组、未透析组、PD组、HD组的Observed species指数和Chao1指数组间比较差异均无统计学意义(P均> 0.05)。与健康对照组比较,未透析组ESRD患者α多样性中的Simpson指数升高(P < 0.008),HD组与PD组间Simpson指数比较差异无统计学意义(P> 0.008),见表2。Unifrac距离矩阵图非加权结果中未透析组、PD组、HD组部分患者与健康对照组空间距离较远,提示组间物种组成结构有差异,但各组之间未见明显分群。未透析组、PD组、HD组患者肠道菌群组内样本较分散,而健康者的样本较集中(图1A),提示患者肠道菌群个体差异较大。基于非加权的PC1主成分和PC2主成分对4组研究对象进行分析,0表示两个微生物群落间OTU的种类一致,采用Bonferroni法校正检验水准后,各组间PC1主成分和PC2主成分两两比较差异均无统计学意义(P均> 0.008),见图1B、C。加权结果中,4组研究对象的菌群均较为分散(图 1D),0则表示群落间OTU的种类和数量均一致,基于加权的PC1主成分分析显示HD组的多样性与ESRD组和PD患者均不同(P均< 0.008),在PC2主成分方面,4组间比较差异无统计学意义(P > 0.05),见图1E、F。
表2 健康对照组、未透析组、PD组、HD组的Observed species 、Chao1、Simpson指数比较 |
| 指 数 | 健康对照组 | 未透析组 | PD组 | HD组 | F/H值 | P值 |
|---|---|---|---|---|---|---|
| Chao1 | 243±25 | 267±92 | 232±51 | 257±67 | 5.927 | 0.115 |
| Observed species | 168±18 | 200±74 | 166±41 | 189±55 | 6.670 | 0.083 |
| Simpson | 0.851(0.796,0.866) | 0.910(0.845,0.923) | 0.882(0.839,0.938) | 0.865(0.806,0.921) | -24.816 | 0.001 |
| 非加权PC1 | -0.058±0.053 | 0.031±0.141 | -0.043±0.082 | 0.027±0.120 | 9.456 | 0.024 |
| 非加权PC2 | 0.046(0.032,0.059) | 0.039(-0.105,0.071) | 0.036(-0.150,0.079) | 0.072(0.056,0.088) | 8.329 | 0.040 |
| 加权PC1 | 0.017±0.155 | -0.037±0.105 | 0.017±0.147 | 0.116±0.140 | 6.979 | <0.001 |
| 加权PC2 | 0.002±0.070 | -0.009±0.066 | 0.007±0.110 | -0.013±0.081 | 0.340 | 0.796 |
三、各组之间肠道菌群组成的差异性分析
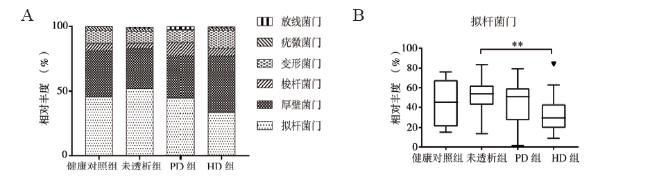
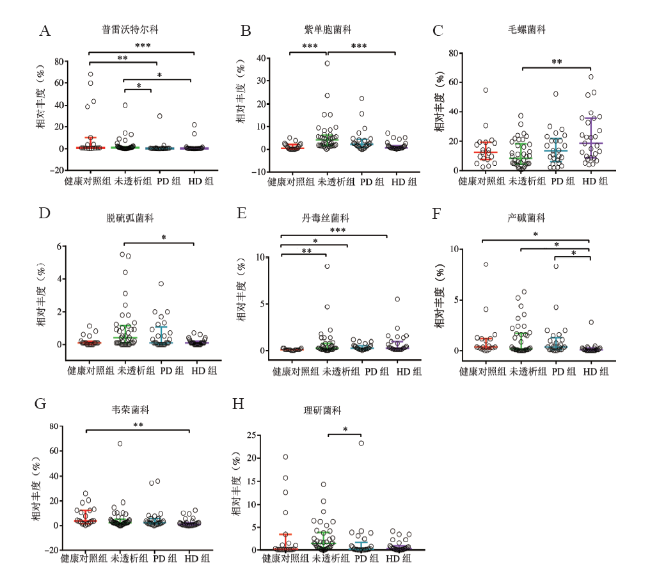
肠道菌群按照门、纲、目、科、属进行分类,与健康对照组比较,未透析组中居前3位的优势菌种为紫单胞菌科(Porphyromonadaceae)、副杆菌属(Parabacteroides)、颤螺旋菌属(Oscillospira);PD组中居前3位的优势菌种为紫单胞菌科、副杆菌属、布劳特菌属(Blautia);HD组中居前3位的优势菌种为瘤胃球菌属(Ruminococcus)、布劳特菌属、粪球菌属(Coprococcus)。4组患者的肠道中均以拟杆菌门(Bacteroidetes)和厚壁菌门(Firmicutes)为主(图2A)。在相对丰度排名前6位的菌门中,与未透析组相比,HD患者肠道中的拟杆菌门比例减少(图2B),其余5个菌门如放线菌门(Actinobacteria)、厚壁菌门、梭杆菌门(Fusobacteria)、变形菌门(Proteobacteria)、疣微菌门(Verrucomicrobia)组间分布均近似。在科水平上,有8个菌科在各组之间比较差异有统计学意义(P < 0.05),无论是与对照组还是未透析组比较,PD组和HD组患者肠道菌群中的普雷沃特尔科(Prevotellaceae)均减少(图3A)。与未透析组比较,HD组毛螺菌科(Lachnospiraceae)比例较高、紫单胞菌科和脱硫弧菌科(Desulfovibrionaceae)比例较低,PD组理研菌科(Rikenellaceae)比例较低(图3B ~ D、H)。与健康对照组比较,未透析组紫单胞菌科比例较高,未透析组、PD组、HD组丹毒丝菌科(Erysipelotrichaceae)比例均较高(图3B、E)。产碱菌科(Alcaligenaceae)和韦荣菌科(Veillonellaceae)在健康对照组、未透析组、PD组中所占的比例相似(P均> 0.05),但在HD组中下降(图3F、G)。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
讨 论
在我国,CKD的发生率逐年上升,多种因素如高血磷、高甲状腺旁腺素和尿蛋白升高等均可以导致肾功能减退[7]。尽管可以利用血液透析或腹膜透析帮助缓解患者的症状,但ESRD患者仍会出现各种并发症,病死率较高。WHO预测,至2030年,每10万人中有14人因CKD死亡,病死率将持续增加[8]。CKD患者除了需要长期接受药物治疗,如抗生素、糖皮质激素或免疫抑制药、钙磷调节药物等,还需严格控制饮食,如避免食用富含钠、钾和磷酸盐的食物等,这些均可影响肠道菌群的定植及生长,易导致心血管疾病、感染、贫血、内环境代谢紊乱等的发生、发展[9]。正常情况下,肠黏膜屏障可阻止有害微生物或毒性代谢产物进入血液循环,肠道菌群能够维持肠道内环境的稳定。肠道菌群失调时,产生多种毒素,并通过受损的肠黏膜上皮进入血循环。
本研究显示,未透析组的肠道菌群α多样性较健康对照组下降,但HD组和PD组的α多样性与未透析组相似。β多样性未见明显分群,但主成分统计分析显示各组间存在差异,未透析组、PD组、HD组的组内差异较大,提示透析可影响ESRD肠道菌群多样性,但未造成严重破坏。Crespo-Salgado等[10]对接受腹膜透析或血液透析治疗的美国中西部儿童和年龄匹配健康儿童的肠道微生物群进行比较,采用腹膜透析Whole tree和Chao1指数评价,结果显示血液透析患儿肠道菌群与健康儿童相似,腹膜透析患儿的α多样性显著下降。另有研究对中国南方的ESRD患者和健康对照者进行肠道菌群测序分析,结果显示两者的α多样性和β多样性相似[11]。由此推测,研究结果不一样可能是受年龄、种族、地域、基础病因等多种因素的影响。
本研究还显示,未透析组肠道中普雷沃特尔科减少,但在PD组和HD组中减少更明显。HD组肠道中毛螺菌科所占的比例比未透析组明显增加。普雷沃特尔科和韦荣氏菌科均可产生短链脂肪酸[12]。拟杆菌门主要产生琥珀酸盐和乙酸盐[13]。短链脂肪酸包括丙酸盐,乙酸盐和丁酸盐,具有维持肠道屏障功能和抗炎作用[14]。腹膜透析和血液透析造成产生短链脂肪酸的菌群减少,易导致肠道屏障的破坏和炎症反应的发生。有研究表明,CKD患者发生心脑血管疾病的风险与毛螺菌科过度生长及普雷沃特尔科的减少有关[3]。上述研究结果均提示,透析患者易发生心脑血管疾病的原因可能与某些肠道菌群的数量变化有关。普雷沃菌属(Prevotella)的下降影响粪便的形成及排空[15]。肠道菌群紊乱易导致便秘,代谢产生的有害物质经肠道上皮吸收增加,促进尿毒症毒素的累积。丹毒丝菌科与结肠癌和胃肠道炎症性疾病相关,还与代谢性疾病相关,在肥胖或高胆固醇血症人群肠道中具有更高比例的丹毒丝菌科[16]。上述研究均提示,ESRD和透析治疗会导致肠道中某些潜在致病菌的增多,疾病发生风险增加。
脱硫弧菌科是变形杆菌的一个亚目,是一种硫酸盐还原微生物,可利用硫酸盐进行无氧呼吸,将其还原为硫化氢,一定量的硫化氢对肠黏膜具有保护性作用[17]。然而,高浓度的硫化氢对结肠收缩性有抑制作用[18,19]。本研究中,未透析组脱硫弧菌科在肠道菌群中所占的比例多于健康对照组。与未透析组相比,HD组脱硫弧菌科减少,与健康对照组相近。此外,在4组研究对象中仅HD组肠道中产碱菌科减少,有报道指,肠道中脱硫弧菌科和产碱菌科可以产生脲酶[11]。脲酶分解尿素所产生的氨,会导致肠道上皮屏障结构和功能的大量破坏,使得肠道源性尿毒症毒素、内毒素、抗原和肠道微生物或其他微生物产物进入循环[5]。有报道紫单胞菌科的富集与腺瘤和癌有关[20]。本研究中,未透析组中肠道中紫单胞菌科增多,而HD组肠道中紫单胞菌科减少。由此推测,血液透析在一定程度上可以改善机体的内环境紊乱,帮助恢复肠道菌群的平衡。
Stadlbauer等[21]研究了3例ESRD患者(15例血液透析,15例腹膜透析)的粪便微生物组成和功能,并与21名健康对照者进行了比较。结果显示,与健康对照者相比,血液透析患者的潜在致病菌增加、有益菌减少,腹膜透析患者的有益菌减少程度较小,并提出了肠道菌群的变化与患者炎症相关,而与肠道通透性无关,肾脏替代治疗是ESRD中生态失调的重要因素。多项研究已经报道过ESRD患者存在肠道菌群失衡,本研究增加了研究对象的样本量,并进行腹膜透析和血液透析患者与未透析的ESRD患者肠道菌群构成的比较,排除了ESRD本身对肠道菌群的影响,进一步明确了透析对ESRD患者的影响,以及血液透析和腹膜透析对肠道菌群改变的不同之处,有助阐明透析造成ESRD患者微量元素及其他临床相关指标改变的原因,为改善ESRD患者的生活质量提供策略。
本研究有一定的局限性。本研究是单中心横断面研究,样本量较小,可能存在选择偏倚。相对于其他物种来说,人类个体间差异较大,易受饮食和药物等多种因素的影响[22]。日后我们将继续追踪未透析的ESRD患者接受透析治疗后肠道菌群的变化,避免个体差异带来的影响。
综上所述,无论是血液透析还是腹膜透析均改变了ESRD患者的肠道菌群组成,ESRD患者肠道菌群的多样性受到了影响,但没有遭到严重破坏。透析治疗在某些程度上增加了一些潜在致病菌,易诱发感染、心血管疾病等并发症,但也能通过透析帮助清除体内毒素,抑制致病菌的生长繁殖和促进有益菌的积累,从而缓解肠道菌群的紊乱。肠道微生物群的改变与透析治疗肾脏疾病是一个相互作用的过程,维持透析患者肠道微生态平衡具有重要作用。